医疗3D打印的春天:医疗器械监督管理规定(试行)
时间:2019-07-05 11:39 来源:南极熊 作者:中国3D打印网 阅读:次
2019年7月4日,南极熊发现国家药监局、国家卫生健康委发布了《定制式医疗器械监督管理规定(试行)的公告(2019年 第53号) 》。

某医疗3D打印人士感叹,“千呼万唤始出来!从2016年11月28日第一次到国家药监局参加定制医疗研讨会到今天,算来已经948天,我的头发都熬白了!”的确,这是中国医疗3D打印领域的一大喜事!每个病人具体的组织器官病灶都是独一无二的,传统的医疗器械一般都是标准化的形状和大小,无法真正贴合每个病人的需求;而3D打印则可以提供快速、相对低成本的器械制造,非常适合在医疗定制上进行应用。广泛而刚性的需求,可以让3D打印定制医疗器械造福国人。但是之前由于没有相关的法律法规,医疗3D打印应用进展缓慢。国内首个获批的金属3D打印植入物,也是靠中央领导人拍板才获得了CFDA三类注册许可的审批。
《定制式医疗器械监督管理规定(试行)》正式发布
为满足临床实践中的罕见特殊个性化需求,规范定制式医疗器械监督管理,保障定制式医疗器械的安全性、有效性,国家药监局和国家卫健委联合发布《关于发布<定制式医疗器械监督管理规定(试行)>的公告》(以下简称《规定》),自2020年1月1日起正式施行。《规定》共分为总则、备案管理、设计加工、使用管理、监督管理和附则六章共35条,明确了定制式医疗器械的定义、备案、设计、加工、使用、监督管理等方面的要求。定制式医疗器械,是指为满足指定患者的罕见特殊病损情况,在我国已上市产品难以满足临床需求的情况下,由医疗器械生产企业基于医疗机构特殊临床需求而设计和生产,用于指定患者的、预期能提高诊疗效果的个性化医疗器械。考虑到定制式医疗器械仅用于特定患者,数量极少,难以通过现行注册管理模式进行注册,《规定》明确对定制式医疗器械实行备案管理,定制式医疗器械生产企业和医疗机构共同作为备案人。为合理控制风险,《规定》对生产、使用定制式医疗器械的生产企业和医疗机构均提出了明确要求,并明确定制式医疗器械不得委托生产。
《规定》同时明确,当定制式医疗器械临床使用病例数及前期研究能够达到上市前审批要求时,应当按照《医疗器械注册管理办法》《体外诊断试剂注册管理办法》规定,申报注册或者办理备案。符合伦理准则且真实、准确、完整、可溯源的临床使用数据,可以作为临床评价资料用于注册申报。《规定》的发布实施,将进一步鼓励定制式医疗器械的创新研发,规范和促进行业的健康发展,满足临床罕见特殊个性化需求,有力保障公众用械安全。

△电子束金属3D打印的钛合金骨科植入物
《定制式医疗器械监督管理规定(试行)》解读
一、关于定义。个性化医疗器械是指医疗器械生产企业根据医疗机构经授权的医务人员提出的临床需求设计和制造的、满足患者个性化要求的医疗器械,分为定制式医疗器械和患者匹配医疗器械。
定制式医疗器械是指为满足指定患者的罕见特殊病损情况,在我国已上市产品难以满足临床需求的情况下,由医疗器械生产企业基于医疗机构特殊临床需求而设计和生产,用于指定患者的、预期能提高诊疗效果的个性化医疗器械。因此,定制式医疗器械具有以下特点:一是用于诊断治疗罕见特殊病损情况,预期使用人数极少,没有足够的人群样本开展临床试验;二是我国已上市产品难以满足临床需求;三是由临床医生提出,为满足特殊临床需求而设计生产;四是用于某一特定患者,预期能提高诊疗效果。
患者匹配医疗器械是指医疗器械生产企业在依据标准规格批量生产医疗器械产品基础上,基于临床需求,按照验证确认的工艺设计和制造的、用于指定患者的个性化医疗器械。患者匹配医疗器械具有以下特点:一是在依据标准规格批量生产医疗器械产品基础上设计生产、匹配患者个性化特点,实质上可以看作标准化产品的特定规格型号;二是其设计生产必须保持在经过验证确认的范围内;三是用于可以进行临床研究的患者人群。如定制式义齿、角膜塑形用硬性透气接触镜、骨科手术导板等。患者匹配医疗器械应当按照《医疗器械注册管理办法》《体外诊断试剂注册管理办法》的规定进行注册或者备案,注册/备案的产品规格型号为所有可能生产的尺寸范围。
二、关于监管方式。考虑到产品特点,定制式医疗器械难以通过现行注册管理模式进行注册,因此对定制式医疗器械实行上市前备案管理。定制式医疗器械生产企业与医疗机构共同作为备案人,在生产、使用定制式医疗器械前应当向医疗器械生产企业所在地(进口产品为代理人所在地)省、自治区、直辖市药品监督管理部门备案。从风险控制的角度出发,定制式医疗器械不得委托生产,备案人应当具备相应条件。当定制式医疗器械生产企业不具备相同类型的依据标准规格批量生产的医疗器械产品的有效注册证或者生产许可证时,或者主要原材料、技术原理、结构组成、关键性能指标及适用范围基本相同的产品已批准注册的,备案自动失效。备案人应当主动取消备案。
定制式医疗器械研制、生产除应当符合医疗器械生产质量管理规范及相关附录要求外,还应当满足特殊要求,包括医工交互的人员、设计开发、质量控制及追溯管理方面的要求。
定制式医疗器械的说明书标签应当体现定制的特点,可以追溯到特定患者。为加强上市后监管,定制式医疗器械的生产和使用实行年度报告制度;对于定制式医疗器械使用及广告、患者信息保护也提出了相应要求。
当定制式医疗器械临床使用病例数及前期研究能够达到上市前审批要求时,相关生产企业应当按照《医疗器械注册管理办法》《体外诊断试剂注册管理办法》规定,申报注册或者办理备案。符合伦理准则且真实、准确、完整、可溯源的临床使用数据,可以作为临床评价资料用于注册申报。如金属3D打印定制式颈椎融合体,在临床应用一定例数、产品基本定型后,可以作为患者匹配医疗器械申报注册。
三、不适用情形。患者匹配医疗器械,符合《医疗器械应急审批程序》有关规定的医疗器械,以及含有药物成分或者细胞、组织等生物活性成分的定制式医疗器械均不适用于本《规定》。
相关阅读
- 国家药监局官方链接http://www.nmpa.gov.cn/WS04/CL2138/338728.html
- 《国家药监局:医用增材制造、人工智能技术医疗器械标准化技术归口单位专家名单》http://www.nanjixiong.com/thread-133104-1-1.html
- 《上海食药监局:3D打印医疗最大困境不是技术,而是法规》http://www.nanjixiong.com/thread-119892-1-1.html
(责任编辑:admin)

 上海器审:金属增材制造定
上海器审:金属增材制造定 工信部等三部门《关于推动
工信部等三部门《关于推动 工信部等三部委发布:推动
工信部等三部委发布:推动 人社部、工信部颁布《增材
人社部、工信部颁布《增材 国务院发展研究中心:增材
国务院发展研究中心:增材 DNV发布了最新版的金属3D
DNV发布了最新版的金属3D最新内容
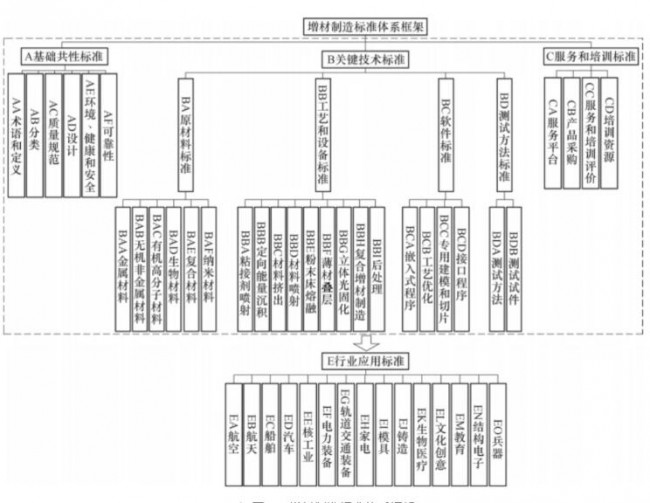 3D打印-增材制造
3D打印-增材制造 《智能制造知识体
《智能制造知识体 2021年中国及31省
2021年中国及31省 全球工程标准协会
全球工程标准协会 毛孔的SLM金属3D
毛孔的SLM金属3D 团体准正式实施!
团体准正式实施!热点内容


